上一次,我们介绍如何根据显著性snp,使用bedtools根据上下游距离,根据gff文件注释基因。
这一次,介绍一下如何根据注释的基因,进行富集分析,主要是看一下GWAS定位的基因有没有某一个趋势,也算是一种验证的方法。比如籽粒大小找到的30个候选基因,如果都与籽粒发育相关的生化途径一致,那就说明找到的都是相关的基因。
之前用于注释基因需要的gff文件:

上面红框中就是基因的名字,这里,我们已经注释到的基因,形成一个txt文件,内容如下:
1. R包依赖
下面先载入需要的R包,如果没有安装,需要安装一下:
library(clusterProfiler)
library(enrichplot)
library(topGO)
library(Rgraphviz)
library(openxlsx)
library(ggplot2)
2. 下载数据库
到Bioconductor中(https://www.bioconductor.org/),检索该物种的数据库:

常见的物种数据库如下:直接在Bioconductor中安装OrgDB的名称就行了。
这里,我们用的是水稻的数据库,名称为:org.Osativa.eg.db
3. 载入数据库和读取基因名文件
载入数据库
library(org.Osativa.eg.db)
db <- org.Osativa.eg.db
organism <- "dosa" # 物种的名称
读取基因型文件
geneid = read.csv("gene_total.txt",header = F)
head(geneid)

4. 将ID匹配GID
将geneID,替换为数据库中的GID
map_id = AnnotationDbi::select(db, keys = geneid, columns=c("GID"), keytype = "RAP")
head(map_id)

5. 对基因列表进行GO注释
GO注释包括:
- MF注释
- CC注释
- BP注释
MF注释:
go_MF =enrichGO(map_id$GID,
OrgDb=db,
keyType = "GID",
ont="MF",
pvalueCutoff=1,
qvalueCutoff=1,
pAdjustMethod="none")
write.xlsx(go_MF,"go_MF.xlsx")
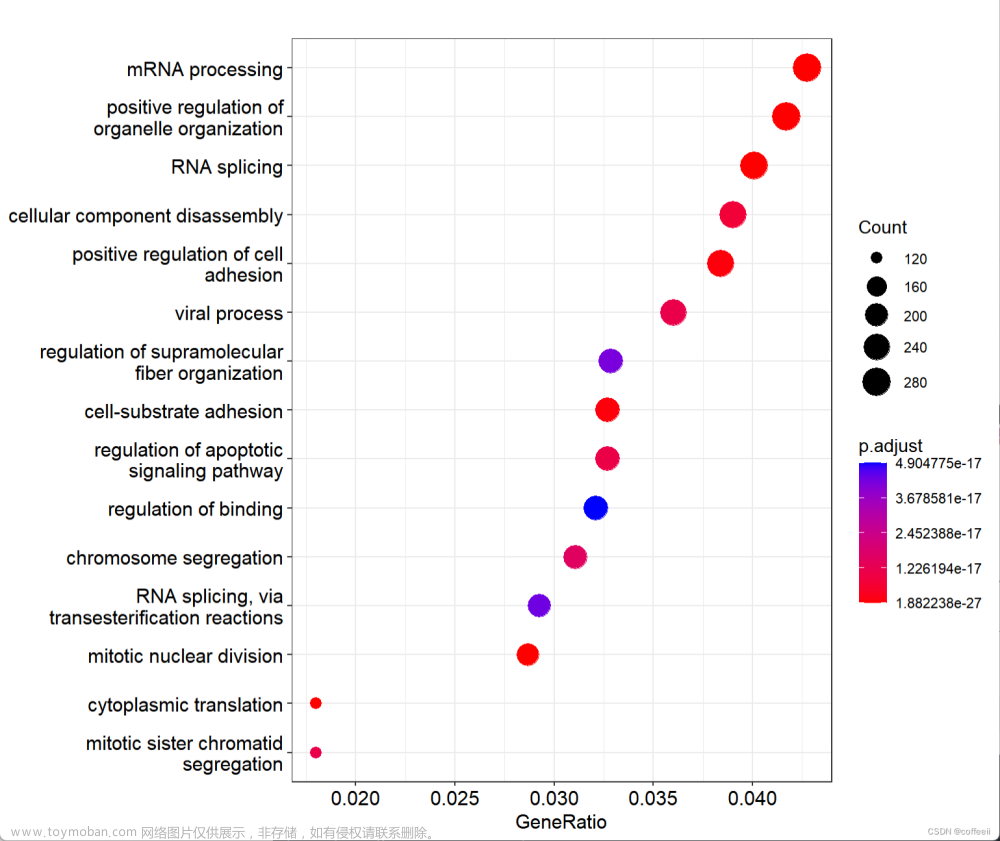
dotplot(go_MF,color="pvalue")
ggsave("go_MF_dotplot.pdf",width=12,height=6)
结果文件:

同样的,CC和BP的GO注释,将ont后面的改为CC和BP即可。
CC的GO注释:
## CC
go_CC =enrichGO(map_id$GID,
OrgDb=db,
keyType = "GID",
ont="CC",
pvalueCutoff=1,
qvalueCutoff=1,
pAdjustMethod="none")
write.xlsx(go_CC,"go_CC.xlsx")
dotplot(go_CC,color="pvalue")
ggsave("go_CC_dotplot.pdf",width=12,height=6)


BP的GO注释:
## BP
go_BP =enrichGO(map_id$GID,
OrgDb=db,
keyType = "GID",
ont="BP",
pvalueCutoff=1,
qvalueCutoff=1,
pAdjustMethod="none")
write.xlsx(go_BP,"go_BP.xlsx")
dotplot(go_BP,color="pvalue")
ggsave("go_BP_dotplot.pdf",width=12,height=6)


其它类型的图:
## 其它类型的图:
barplot(go_BP)
heatplot(go_BP)


6. KEGG富集分析
把基因型的ID后面加上“-01”,并且把g变为t
rap_id <- paste0(geneid, "-01")
rap_id <- gsub("g","t",rap_id)
head(rap_id)

富集分析:
geneid = read.csv("a1.txt",header = F)$V1
rap_id <- paste0(geneid, "-01")
rap_id <- gsub("g","t",rap_id)
head(rap_id)
kegg <- enrichKEGG(
gene = rap_id, #基因列表文件中的基因名称
keyType = 'kegg',
organism = 'dosa',
pAdjustMethod = 'fdr', #指定 p 值校正方法
pvalueCutoff = 1,
qvalueCutoff = 1)
运行日志:
作图:
barplot(kegg)
dotplot(kegg)
 文章来源:https://www.toymoban.com/news/detail-452677.html
文章来源:https://www.toymoban.com/news/detail-452677.html

 文章来源地址https://www.toymoban.com/news/detail-452677.html
文章来源地址https://www.toymoban.com/news/detail-452677.html
到了这里,关于GWAS分析中的GO和KEGG富集分析的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!