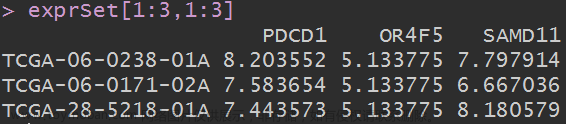
geo读取表达矩阵 RNA-seq R语言
方法一:1.从geo页面直接下载表达矩阵,然后通过r读取表达矩阵
2.利用getgeo函数读取表达矩阵
3.利用geo自带的geo2r,调整p值为1,获取探针和基因名的对应关系
1
#http://zouyawen.top/2020/10/09/%E8%BD%AC%E5%BD%95%E7%BB%844/
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE150425_a549_kras_organoid/")
expr.df=read.csv("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE150425_a549_kras_organoid/GSE150425_Cufflinks_Gene_Counts/GSE150425_Cufflinks_Gene_Counts.csv",
header=T,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(expr.df)
head(expr.df)
expr.df[expr.df$Gene_ID %in% 'RETN',]
library(dplyr)
colnames(expr.df)[1]='Gene_Symbol'
expr.df[expr.df$Gene_Symbol=='Retn'|
expr.df$Gene_Symbol=='Retnlb'|
expr.df$Gene_Symbol=='Jchain'|
expr.df$Gene_Symbol=='Igha1'|
expr.df$Gene_Symbol=='Pdk4'|
expr.df$Gene_Symbol=='Actb',]

#http://zouyawen.top/2020/10/09/%E8%BD%AC%E5%BD%95%E7%BB%844/
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE133037_a549_transfection 0f runx__tgfb/")
expr.df=read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE133037_a549_transfection 0f runx__tgfb/GSE133037_VK-Rx3.TGFb_FPKM/GSE133037_VK.Rx3.TGFb_FPKM.txt",
header=T,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(expr.df)
#colnames(expr.df)=expr.df[1,]
input=expr.df
head(input)
library(dplyr)
input=input[,!(grepl(colnames(input),pattern = 'Chr')|
grepl(colnames(input),pattern = 'Start')|
grepl(colnames(input),pattern = 'End')|
grepl(colnames(input),pattern = 'Strand'))]
head(input)
colnames(input)[1]='Gene.ID'
colnames(input)[2]='Gene.Name'
head(input)
mydata=input %>% filter(Gene.Name=='RETNLB'|Gene.Name=='ACTB'|Gene.ID=='1053'|
Gene.Name=='56729'|Gene.ID=='3493'|
Gene.ID=='3512'|Gene.ID=='3934')
mydata
dim(input)
多个组别 合并 id转化
#http://zouyawen.top/2020/10/09/%E8%BD%AC%E5%BD%95%E7%BB%844/
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/GSE135402_RAW/GSM4007697_A549-2/")
expr_SRR957678=read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/GSE135402_RAW/GSM4007697_A549-2/GSM4007696_A549-1.htseq.count.txt",
header=FALSE,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
expr_SRR957677=read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/GSE135402_RAW/GSM4007697_A549-2/GSM4007697_A549-2.htseq.count.txt",
header=FALSE,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
expr_SRR957679 =read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/GSE135402_RAW/GSM4007699_TD-2/GSM4007698_TD-1.htseq.count.txt",
header=FALSE,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
expr_SRR957680 =read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/GSE135402_RAW/GSM4007699_TD-2/GSM4007699_TD-2.htseq.count.txt",
header=FALSE,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(expr_SRR957677)
head(expr_SRR957678)
#先对照和实验组分别合并到一个data.frame再合并为一个总的数据框
control <- merge(expr_SRR957677,expr_SRR957678,by="V1")#by="gene_id"按照gene_id这列合并
head(control)
treat <- merge(expr_SRR957679,expr_SRR957680,by="V1")
head(treat)
raw_count <- merge(control,treat,by="V1")
head(raw_count)
colnames(raw_count)=c('gene_id','a5491','a5492','td1','td2')
raw_count <-raw_count[-1:-5,]#删掉前五行,由上一步结果可以看出前五行gene_id有问题
head(raw_count)
2#.去除gene_id名字中的小数点
#EBI数据库中无法搜到ENSG00000000003.10这样的基因,只能是整数ENSG00000000003才能进行id转换
library(stringr)
ENSEMBL <- raw_count$gene_id
head(ENSEMBL)
ENSEMBL <- str_split_fixed(ENSEMBL,'[.]',2)[,1]#将小数点及后面的数字去掉,ensembl_gene_id是整数
head(ENSEMBL)
rownames(raw_count) <- ENSEMBL#将去除小数点后的ensembl_gene_id作为行名后期方便绘图
raw_count$ensembl_gene_id <- ENSEMBL#新建ensembl_gene_id列便于合并
head(raw_count)
3.#对基因进行注释,获取gene_symbol
#bioMart包是一个连接bioMart数据库的R语言接口,能通过这个软件包自由连接到bioMart数据库
# BiocManager::install('biomaRt')
library(biomaRt)
#显示一下能连接的数据库
listMarts()
listMarts()
#用useMart函数选定数据库
plant<-useMart("ensembl")
#用listDatasets()函数显示当前数据库所含的基因组注释,我们要获取的基因注释的基因是人类基因,所以选择hsapiens_gene_ensembl
listDatasets(plant)
grep(listDatasets(plant)[,1],pattern = 'hsa')
#选定ensembl数据库中的hsapiens_gene_ensembl基因组
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
#选定我们需要获得的注释类型
#用lsitFilters()函数查看可选择的类型,选定要获取的注释类型,以及已知注释的类型用lsitFilters()函数查看可选择的类型,选定要获取的注释类型,以及已知注释的类型
listFilters(mart)
#这里我们选择这些要获得数值的类型ensembl_gene_id ,hgnc_symbol chromosome_name start_position end_position band我们已知的类型是ensembl_gene_id选择好数据库,基因组,要获得的注释类型,和已知的注释类型,就可以开始获取注释了
#用getBM()函数获取注释
hg_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"), filters= 'ensembl_gene_id', values = ENSEMBL, mart = mart)
#attributers()里面的值为我们要获取的注释类型ensembl_gene_id ,hgnc_symbol chromosome_name start_position end_position band
#filters()里面的值为我们已知的注释类型ensembl_gene_id
#values= 这个值就是我们已知的注释类型的数据,把上面我们通过数据处理得到的ensembl基因序号作为ensembl_gene_id 的值
#mart= 这个值是我们所选定的数据库的基因组
head(hg_symbols)
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb")
save(mart, file = "mart.RData")#将mart变量保存为mart.RData
save(hg_symbols, file = "hg_symbols.RData")#将hg_symbols变量保存为hg_symbols,biomart包不太稳定,有时连不上
4.#将合并后的表达数据框raw_count与注释得到的hg_symbols整合
read_count <- merge(raw_count,hg_symbols,by="ensembl_gene_id")#将raw_count与注释后得到的hg_symbols合并
head(read_count)
openxlsx::write.xlsx(read_count,file = "readcount_all.xlsx")
input =openxlsx::read.xlsx("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE135402_A549_TD_tgfb/readcount_all.xlsx")
library(dplyr)
input=input[,!(grepl(colnames(input),pattern = 'Chr')|
grepl(colnames(input),pattern = 'Start')|
grepl(colnames(input),pattern = 'End')|
grepl(colnames(input),pattern = 'Strand'))]
head(input)
colnames(input)[1]='Gene.ID'
colnames(input)[7]='Gene.Name'
head(input)
mydata=input %>% filter(Gene.Name=='RETNLB'|Gene.Name=='ACTB'|Gene.ID=='ENSG00000104918'|
Gene.Name=='RETN'|Gene.ID=='ENSG00000132465'|
Gene.ID=='ENSG00000211895'|Gene.ID=='ENSG00000092067')
mydata
dim(input)
下载表达矩阵和getgeo函数联合使用文章来源:https://www.toymoban.com/news/detail-615344.html
#http://zouyawen.top/2020/10/09/%E8%BD%AC%E5%BD%95%E7%BB%844/
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE114761_various_A549_TGFB/")
expr.df=read.table("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE114761_various_A549_TGFB/geo2r.tsv",
header=T,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(expr.df)
expr.df[1:10,]
#colnames(expr.df)=expr.df[1,]
input=expr.df
head(input)
library(dplyr)
input=input[,!(grepl(colnames(input),pattern = 'Chr')|
grepl(colnames(input),pattern = 'Start')|
grepl(colnames(input),pattern = 'End')|
grepl(colnames(input),pattern = 'Strand'))]
head(input)
grep(input$Gene.title,pattern = 'RETN')
input[grep(input$Gene.symbol,pattern = 'RETN'),]
input[grep(input$Gene.symbol,pattern = 'IGHA1'),]
input[grep(input$Gene.title,pattern = 'immunoglobulin heavy'),]
library(GEOquery)
f='GSE114761_eSet.Rdata'
if(!file.exists(f)){
gset <- getGEO('GSE114761', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file=f) ## 保存到本地
}
getwd()
load('GSE114761_eSet.Rdata') ## 载入数据
class(gset) #查看数据类型
length(gset) #
class(gset[[1]])
gset
exprs(gset[[1]])
gset[[1]]
a=exprs(gset[[1]])
head(a)
a['220570_at',]
a[rownames(a)=='220570_at',]
input[grep(input$Gene.symbol,pattern = 'IGHA1'),][,1]
a[rownames(a) %in% input[grep(input$Gene.symbol,pattern = 'IGHA1'),][,1],]
a[rownames(a) %in% input[grep(input$Gene.title,pattern = 'IGCJ'),][,1],]
myexpression=as.data.frame(a)
head(myexpression)
myexpression$probe_id=rownames(myexpression)
head(myexpression)
head(input)
input$probe_id=input$ID
head(input)
mydata=merge(myexpression,input,by='probe_id')
head(mydata)
colnames(a)
boxplot(mydata[,2:42])
getwd()
save(mydata,file = "G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE114761_various_A549_TGFB/myexpression_matrix.rds")
load("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE114761_various_A549_TGFB/myexpression_matrix.rds")
colnames(mydata)
a=read.table(file = 'clipboard')
head(a)
a=a[-1,]
a
colnames(mydata)[2:43]=a$V2
head(mydata)
mydata[mydata$Gene.symbol=='RETN'|
mydata$Gene.symbol=='IGHA1'|
mydata$Gene.symbol=='RETNLB',]
读取excel表达矩阵文章来源地址https://www.toymoban.com/news/detail-615344.html
getwd()
setwd("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE164160_CMT64_TGFB_Alveogenic lung carcinoma/")
syndecan_fibrosis=openxlsx::read.xlsx("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE164160_CMT64_TGFB_Alveogenic lung carcinoma/GSE164160_RAW/GSM4998575_TGFb_1.clean_trimmed_GE_.xlsx",
colNames = T)
head(syndecan_fibrosis)
input=syndecan_fibrosis
library(dplyr)
input=input[,!(grepl(colnames(input),pattern = 'Region')|
grepl(colnames(input),pattern = 'Start')|
grepl(colnames(input),pattern = 'End')|
grepl(colnames(input),pattern = 'Strand'))]
head(input)
mydata=input %>% filter(Name=='Regnla'|Name=='Actb'|ENSEMBL=='ENSMUSG00000061100'|ENSEMBL=='ENSMUSG00000015839'|
Name=='Retnlg'|ENSEMBL=='ENSMUSG00000052435'|
ENSEMBL=='ENSMUSG00000095079'|ENSEMBL=='ENSMUSG00000067149')
mydata
dim(input)
#################################33
syndecan_fibrosis=openxlsx::read.xlsx("G:/silicosis/geo/GSE103548_rna-seq_llc_mle-12/GSE164160_CMT64_TGFB_Alveogenic lung carcinoma/GSE164160_RAW/GSM4998576_TNFa_1.clean_trimmed_GE_.xlsx",
colNames = T)
head(syndecan_fibrosis)
input=syndecan_fibrosis
library(dplyr)
input=input[,!(grepl(colnames(input),pattern = 'Region')|
grepl(colnames(input),pattern = 'Start')|
grepl(colnames(input),pattern = 'End')|
grepl(colnames(input),pattern = 'Strand'))]
head(input)
mydata=input %>% filter(Name=='Regnla'|Name=='Actb'|ENSEMBL=='ENSMUSG00000061100'|ENSEMBL=='ENSMUSG00000015839'|
Name=='Retnlg'|ENSEMBL=='ENSMUSG00000052435'|
ENSEMBL=='ENSMUSG00000095079'|ENSEMBL=='ENSMUSG00000067149')
mydata
dim(input)
到了这里,关于geo读取表达矩阵 RNA-seq R语言部分(表达矩阵合并及id转换)的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!