#因为之前的命令调用GDCquery_Maf 发现用不了
#故找到了一些其他的方法,并且自己试着将其弄成了一个表达矩阵。
#代码如下
#1、下载加载相应的包
install.packages("pacman")
library(pacman)
p_load(TCGAbiolinks,DT,tidyverse)
BiocManager::install("TCGAbiolinks")
library(TCGAbiolinks)
library(tidyverse)
BiocManager::install("maftools")
library(maftools)
library(dplyr)
#
TCGAbiolinks:::getProjectSummary("TCGA-STAD")
#建立查询
raw<-GDCquery(
project= "TCGA-STAD",
data.category = "Simple Nucleotide Variation",
access = "open",
legacy = FALSE,
data.type= "Masked Somatic Mutation",
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking"
)
#下载数据
GDCdownload(raw)
#获取数据
maf<- GDCprepare(raw)
#读取数据
maf<-maf %>% maftools::read.maf()
#表格化数据,数据会以表格形式出现在右下角viewer里
datatable(getSampleSummary(maf),
filter = 'top',
options = list(scrollX = TRUE , keys = TRUE,
pageLength = 5),
rownames = FALSE)
#(以上代码是看b站一个视频来的,但是我需要这个表格,根据自己需求绘制相应的样本的TMB瀑布图,比较两个组之间的差异情况)
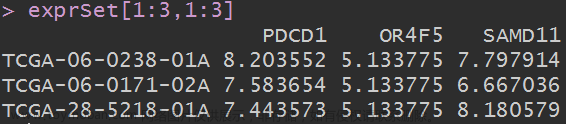
#把突变数据提取成为一个表达矩阵
a<-datatable(getSampleSummary(maf),
filter = 'top',
options = list(scrollX = TRUE , keys = TRUE,
pageLength = 5),
rownames = FALSE)
#观察列表a的结构,发现突变数据表达矩阵在a的x中的data中,提取出来。
STADTMB<-a$x$data
View(STADTMB)

文章来源:https://www.toymoban.com/news/detail-664995.html
#这样就生成了一个和GDCquery_Maf提取出来的合并了的体细胞突变数据了(应该是,自己也没用GDCquery_Maf 提取出来过。总之可以用来做后续的分析:比如比较TCGA中构建出来的风险模型,高低风险组之间的TMB情况)文章来源地址https://www.toymoban.com/news/detail-664995.html
到了这里,关于提取TCGA 中体细胞突变数据的表达矩阵的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!