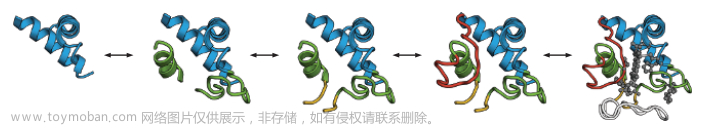
研究背景:
- 病毒感染可以导致多种组织特异性损伤,所以 virus-host PPIs 的预测有助于新的治疗方法的研究;
- 目前已有的一些 virus-host PPIs 鉴定或预测方法效果有限(传统实验方法费时费力、计算方法要么基于蛋白结构或基因,要么基于手动特征工程的机器学习);
- DL在PPIs预测中的应用愈加广泛,包括特征嵌入、autoencoder、LSTM等,而最近几年基于NLP领域的一些基于迁移学习的方法、基于 transformer 的预训练模型的应用等在 PPIs 预测中展现了更好的表现;
- 本文中,作者提出了一个基于 ProtBERT 模型的深度学习方法,名为 STEP(据作者所知,这是第一个用预训练 transformer 模型获取序列嵌入特征用于 PPIs 预测的方法);
数据集构成:

| 数据集 | 构成 |
|---|---|
| Tsukiyama | 22,383 positive PPIs (5,882 human proteins & 996 virus proteins) |
| Guo | 5943 positive PPIs |
| Sun | 36,545 positive PPIs, 36,323 negative PPIs |
- Tsukiyama 的数据集是 human-virus PPIs,正负样本数目比为1:10,负样本构造方法是:dissimilarity-based negative sampling method。此外,将整个数据集中的20%取出作为独立的验证集 (independent test data);
- Guo 的数据集是 Yeast PPIs,其中负样本PPIs的数目和正样本PPIs一样,构建方法有三种:1). 正样本蛋白随机组对;2). 不同亚细胞定位的蛋白进行组对;3). 利用人为构造(将已有蛋白的序列进行打乱)的蛋白序列进行组对;
- Sun 的数据集是 Human PPIs,从 HPRD 数据库中整理得到的,负样本构建方法是 “不同亚细胞定位随机组对” 和 “Negatome database 中的非互作蛋白”;
研究思路和方法:

代码:https://github.com/SCAI-BIO/STEP
从示意图中可以看出,STEP方法的整体结构是很简单的,所以根据上述示意图对主要代码进行简述(代码主要来自src/modeling/ProtBertPPIModel.py):
1. __single_step()方法从宏观上规定了STEP运行过程:

上面截图为ProtBertPPIModel.py的314行-327行,这段代码规定了STEP的运行顺序,其中我觉得比较重要的点我用红线标了出来:
- 该段代码定义了一个
__single_step(self, batch)的方法,其输入是batch,根据315行可以确定batch是由inputs_A, input_B, targets三部分构成的。 - 之后的316和317行表示将
inputs_A和inputs_B输入到self.forward()方法中得到model_out_A和model_out_B,从这一步可以推测出inputs_A是由input_ids, token_type_ids, attention_mask构成。 -
self.forward()的输出作为下一步self.classifier()方法的输入,之后得到classifier_output(即预测输出),之后再利用self.loss_bce_with_integrated_sigmoid()方法计算损失,最终__signle_step()方法返回(loss, trues, preds)(即损失值、真实标签和预测值)。
self.forward()方法定义了ProtBERT模型如何对输入的蛋白序列进行编码的:

首先蛋白A和蛋白B的序列由氨基酸构成的字符串,不能直接输入到神经网络中进行训练,需要将需要将字符串映射为数值型数据。这一步就是干这个事的,也就是用预训练的ProtBERT模型将蛋白质序列进行向量化表示。
-
input_ids, token_type_ids, attention_mask输入self.ProtBertBFD()方法之后得到word_embeddings,之后通过self.pool_strategy()方法对word_embedding进行池化操作,而这个self.pool_strategy()(如下图所示),这里的features指的就是{"token_embeddings": word_embeddings, "cls_token_embeddings": word_embeddings[:, 0], "attention_mask": attention_mask},而self.pool_strategy()的输出output_vectors则计算了三种情况下的池化结果。

- 这里存在的疑问是
input_ids, token_type_ids, attention_mask究竟指的是什么?
根据src/data/VirHostNetDataset.py中(下图所示)可以看出,input_ids, token_type_ids, attention_mask是由self.tokenizer()方法得到的,而self.tokenizer()方法指的是预训练模型Roslab/prot_bert_bfd中的tokenizer,这三个数据可以从tokenizer中得到(可见 https://huggingface.co/Rostlab/prot_bert_bfd)。

self.ProtBertBFD()加载预训练PortBERT模型:

正如上面所述,预训练模型ProtBERT可以直接从 hugging face 上下载得到,通过BertModel.from_pretrained()方法进行加载即可(红框所注部分)。
self.classifier()对蛋白A和蛋白B特征进行哈达玛积,并进行预测分类:

将通过self.forward()方法得到的蛋白A和蛋白B的特征进行哈达玛积,并将结果输入到self.classification_head()方法中即可得到预测结果(其中self.classification_head()方法在上面的__build_model()方法中)。
大概情况就是这样(有错之处,还请指出,及时更改),其他细节详见代码。
实验结果及讨论:
1. Comparative evaluation of STEP with state-of-the-art work:
| 方法 | 特征+模型 |
|---|---|
| Tsukiyama (2021) | word2vec sequence embedding + LSTM-PHV Siamese model (5-fold-cv) |
| Yang (2019) | doc2vec + RF classifier |
| Guo (2008) | auto covariance + SVM (5-fold-cv) |
| Sun (2017) | AC + CT + autoencoder (10-fold-cv) |
| Chen (2019) | Siamese residual RCNN (5-fold-cv) |
| STEP (2022) | ProtBERT + Siamese Neural Network |
1.1 PPIs 预测任务上各方法的预测表现:

1.2 在 PPIs 互作类型和结合亲和力任务上,各方法的预测表现:
- PPIs 互作类型预测:
数据集:SHS27k dataset(由Chen对STRING数据库整理得到),包括 26,944 PPIs,涉及7种互作类型: activation (16.70%), binding (16.70%), catalysis (16.70%), expression (5.84%), inhibition (16.70%), post-translational modification (ptmod; 10.66%), and reaction (16.70%)。- PPIs 结合亲和力预测:
数据来自 SKEMPI 数据库,包括 2,792 突变蛋白复合物的结合亲和力(参考Chen的方法对数据集进行了处理)。- 模型修改:
1). 对于PPIs预测任务(多分类任务),将 bottleneck classification head 替换为三个一样的线性层(dropout和ReLU不变),将损失函数换成 cross-entropy,sigmoid 激活函数换成 Softmax。
2). 对于PPIs 结合亲和力预测(回归问题),将损失函数替换成 mean squared error loss,并将预测值缩放到0-1之间。- 做10-fold-cross validation。

1.3 结论1:
- Table1 demonstrated at least state-of-the-art performance of STEP.
- STEP compared on exactly the same data published by Tsukiyama performs similar to their LSTM-PHV method and better than the approach by Yang.
- TableS4, we also evaluated our STEP architecture on two additional tasks, namely, PPI type prediction and a PPI binding affinity estimation using the data and the CV setup provided by Chen. For both tasks, we reached at least state-of-the-art per- formances with our approach.
2. Prediction of JCV major capsid protein VP1 interactions:
- We split the brain tissue-specific interactome dataset including all positive and pseudo-negative interactions into training (60%), validation (20%), and test (20%) datasets.
- After tuning on the validation set, we used our best model to make predictions on the hold-out test set.
- 之所以用 brain tissue-specific interactome 的数据,是因为 JCV 可以透过血脑屏障入脑。
2.1 超参数优化(模型微调):

2.2 STEP-Brain对于脑组织特异性互作蛋白的预测表现:

2.3 STEP-Brain对于JCV major capsid protein VP1 互作蛋白的预测结果(top10):
We used this STEP-brain model to predict interactions of the JCV major capsid protein VP1 with all human receptors.

2.4 JCV major capsid protein VP1 被预测的互作蛋白富集分析结果:

Altogether, we observed a strong enrichment of VP1 interactions predicted with olfactory, serotonin, amine, taste, and acetylcholine receptors.
3. Prediction of SARS-CoV-2 spike glycoprotein interactions:
3.1 训练思路:
We performed a nested CV procedure on the given SARS-CoV-2 interactions dataset. We used five outer and five inner loops to validate the generalization performance and while performing the hyperparameter optimization in the inner loop. In each outer run, we created a stratified split of the interactome into train (4/5) and test (1/5) datasets. In the nested run, we further split the outer train dataset into train (1/5) and validation (1/5) datasets, which were used to optimize the hyperparameters of the model using the respective training data.
关于 Nested Cross Validation 的示意图(图片来自网络):

3.2 超参数优化:

3.3 STEP-virus-host model 的 Nested CV 测试结果:

 文章来源:https://www.toymoban.com/news/detail-679939.html
文章来源:https://www.toymoban.com/news/detail-679939.html
3.4 STEP-virus-host model 预测SARS-CoV-2 spike 蛋白的人类受体结果:
STEP-virus-host model obtained from the best outer fold to predict interactions of the SARS-CoV-2 spike pro- tein (alpha, delta, and omicron variants) with all human receptors that were not already contained in VirHostNet.
文章来源地址https://www.toymoban.com/news/detail-679939.html
- For all virus variants the sigma intracellular receptor 2 (GeneCards:TMEM97; UniProt:Q5BJF2) was the only one predicted with an outstanding high probability (of >70% in all cases).
- The sigma 1 and 2 receptors are thought to play a role in regulating cell survival, morphology, and differentiation.
- In addition, the sigma receptors have been proposed to be involved in the neuronal transmission of SARS- CoV-2. They have been suggested as targets for therapeutic intervention.
- Our results suggest that the antiviral effect observed in cell lines treated with sigma receptor binding ligands might be due to a modulated binding of the spike protein, thus inhibiting virus entry into cells.
4. 讨论:
- 利用预训练ProtBERT和Siamese neural network架构仅根据蛋白质以及序列来预测 PPIs,结果表明该方法(STEP)比之前的基于LSTM等原理的方法效果更优;
- 通过将STEP进行超参数优化得到的模型可以很好地预测脑组织特异性PPIs以及virus-host PPIs的预测;
- 微调的模型 STEP-Brain 和 STEP-virus-host 可分别用于预测 JCV major capsid protein VP1 互作蛋白以及 SARS-CoV-2 spike glycoprotein 互作受体;
- 作者首次提出将预训练大模型用于PPIs预测,意义还是很重大的。但是整体上来看,尽管模型比较简单,但是对计算资源的要求很高,(每一次微调需要 2xA100GPU with VMEM of 32GB,尽管可以并行,但是微调116次,作者用了10days的时间);
【本文章给我的启发就是,没有足够的计算资源,大模型还是不要搞得好😮💨】
到了这里,关于利用深度蛋白质序列嵌入方法通过 Siamese neural network 对 virus-host PPIs 进行精准预测【Patterns,2022】的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!