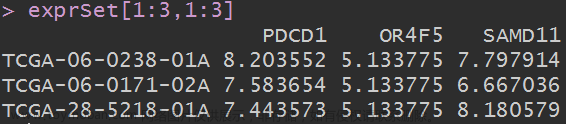
上节,我们使用结核病基因数据,做了一个数据预处理的实操案例。例子中结核类型,包括结核,潜隐进展,对照和潜隐,四个类别。本节延续上个数据,进行了差异分析。
差异分析 计算差异指标step12
加载数据
load("dataset_TB_LTBI_step8.Rdata")构建差异比较矩阵
#样本列表
group_list=group_data_TB_LTBI$group_more
#构建分组
design=model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
#head(dataset_TB_LTBI)
row.names(design)=colnames(dataset_TB_LTBI)
design #得到分组矩阵:0代表不是,1代表是
#str(design)
library(limma)
##差异比较矩阵
contrast_matrix=makeContrasts(paste0(c('LTBI','TB'),collapse = '-'),levels = design)计算差异基因指标
#step:lmFit
fit=lmFit(dataset_TB_LTBI,design)
fit2=contrasts.fit(fit,contrast_matrix)
#step:eBayes
fit3=eBayes(fit2)
#step3:topTable
tempoutput=topTable(fit3,coef = 1,n=Inf)
DEG_M=na.omit(tempoutput) #得到差异分析矩阵,重点看logFC和P值
head(DEG_M) #查看数据
'''
logFC AveExpr t P.Value adj.P.Val B
ASPHD2 -1.452777 8.415563 -12.38370 5.885193e-22 5.868863e-18 39.30255
C1QC -3.978887 5.971935 -12.34993 6.954041e-22 5.868863e-18 39.14037
GBP1P1 -4.075057 5.607978 -12.24397 1.174622e-21 6.608814e-18 38.63087
GBP6 -3.225604 4.393248 -11.93968 5.320543e-21 1.692866e-17 37.16200
SDC3 -2.374911 7.388880 -11.92896 5.612049e-21 1.692866e-17 37.11012
LHFPL2 -1.705514 8.411180 -11.91494 6.017652e-21 1.692866e-17 37.04225
'''#绘制前40个基因在不同样本之间的热图
library(pheatmap)
#绘制前40个基因在不同样本之间的热图
f40_gene=head(rownames(DEG_M),40)
f40_subset_matrix=dataset_TB_LTBI[f40_gene,]
head(f40_subset_matrix)
f40_subset_matrixx=t(scale(t(f40_subset_matrix))) #数据标准化。。。数据标准化和归一化的区别:平移和压缩
pheatmap(f40_subset_matrixx) #出图差异分析 结果过滤筛选step13
res = DEG[,c("logFC","P.Value","adj.P.Val")]
colnames(res)<-c("logFC","PValue","padj")
colnames(res)
library(dplyr)
FC_filter =0.585
P_filter=0.05
all_diff =res %>% filter(abs(logFC)>FC_filter) %>% filter(padj<P_filter)
res$id = rownames(res)
res=select(res,id,everything())
#write.table(res,'all_diff.txt',sep='\t',quote=F)
up_diff=res %>% filter(logFC>FC_filter) %>% filter(padj<P_filter)
up_diff$id = rownames(up_diff)
up_diff=select(up_diff,id,everything())
#write.table(up_diff,'up_diff.txt',sep='\t',quote=F)
down_diff=res %>% filter(logFC< -FC_filter ) %>% filter(padj<P_filter)
down_diff$id = rownames(down_diff)
down_diff=select(down_diff,id,everything())
#write.table(down_diff,'down_diff.txt',sep='\t',quote=F)
group_data_clean <-function(data){
# colnames(data)[c(9,10,11)] =c("logFC","PValue","padj")
data[which(data$padj %in% NA),'sig'] <- 'no diff'
data[which(data$logFC >= FC_filter & data$padj < 0.05),'sig'] <- 'up'
data[which(data$logFC <= -FC_filter & data$padj < 0.05),'sig'] <- 'down'
data[which(abs(data$logFC) < FC_filter | data$padj >= 0.05),'sig'] <- 'no diff'
cat(" 上调",nrow(data[data$sig %in% "up", ]))
cat(" 下调",nrow(data[data$sig %in% "down", ]))
cat(" no fiff",nrow(data[data$sig %in% "no diff", ]))
# filter_data = subset(data, data$sig == 'up' | data$sig == 'down')
# filter_data$Geneid <- rownames(filter_data)
return(data)
}
limma_clean_res = group_data_clean(res)
#上调 1381 下调 1432 no fiff 14066
rownames(all_diff)
dataset_TB_LTBI_DEG = dataset_TB_LTBI[rownames(all_diff),]
dim(dataset_TB_LTBI_DEG) #[1] 2813 102
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(DEG,res,all_diff,limma_clean_res,dataset_TB_LTBI_DEG,file = "DEG_TB_LTBI_step13.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
差异分析 绘制火山图step14
library(ggplot2)
data <- limma_clean_res
#################
# ggplot2绘制火山图
data$label <- c(rownames(data)[1:10],rep(NA,nrow(data) - 10))
#sizeGrWindow(12, 9)
pdf(file="差异基因火山图step14.pdf", width = 9, height = 6)
ggplot(data,aes(logFC,-log10(PValue),color = sig)) +
xlab("log2FC") +
geom_point(size = 0.6) +
scale_color_manual(values=c("#00AFBB","#999999","#FC4E07")) +
geom_vline(xintercept = c(-1,1), linetype ="dashed") +
geom_hline(yintercept = -log10(0.05), linetype ="dashed") +
theme(title = element_text(size = 15), text = element_text(size = 15)) +
theme_classic() +
geom_text(aes(label = label),size = 3, vjust = 1,hjust = -0.1)
dev.off()
 文章来源:https://www.toymoban.com/news/detail-725474.html
文章来源:https://www.toymoban.com/news/detail-725474.html
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(DEG,res,all_diff,limma_clean_res,dataset_TB_LTBI_DEG,file = "DEG_TB_LTBI_step13.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
差异基因分析完毕,下面我们可以观察一下,这些基因富集在哪些通路之上。文章来源地址https://www.toymoban.com/news/detail-725474.html
到了这里,关于GEO生信数据挖掘(七)差异基因分析的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!