前面我们简单介绍过ggplot2画KEGG富集柱形图,其实GO富集结果的展示相对于KEGG来说要复杂一点点,因为GO又进一步可以划分成三个类。
BP:biological process,生物学过程。
MF:molecular function,分子功能。
CC:cellular component, 细胞成分。
因此在画图的时候,我们需要将这三类给区分开来。下面分别用了三种不同的方式来展示GO富集分析的结果。

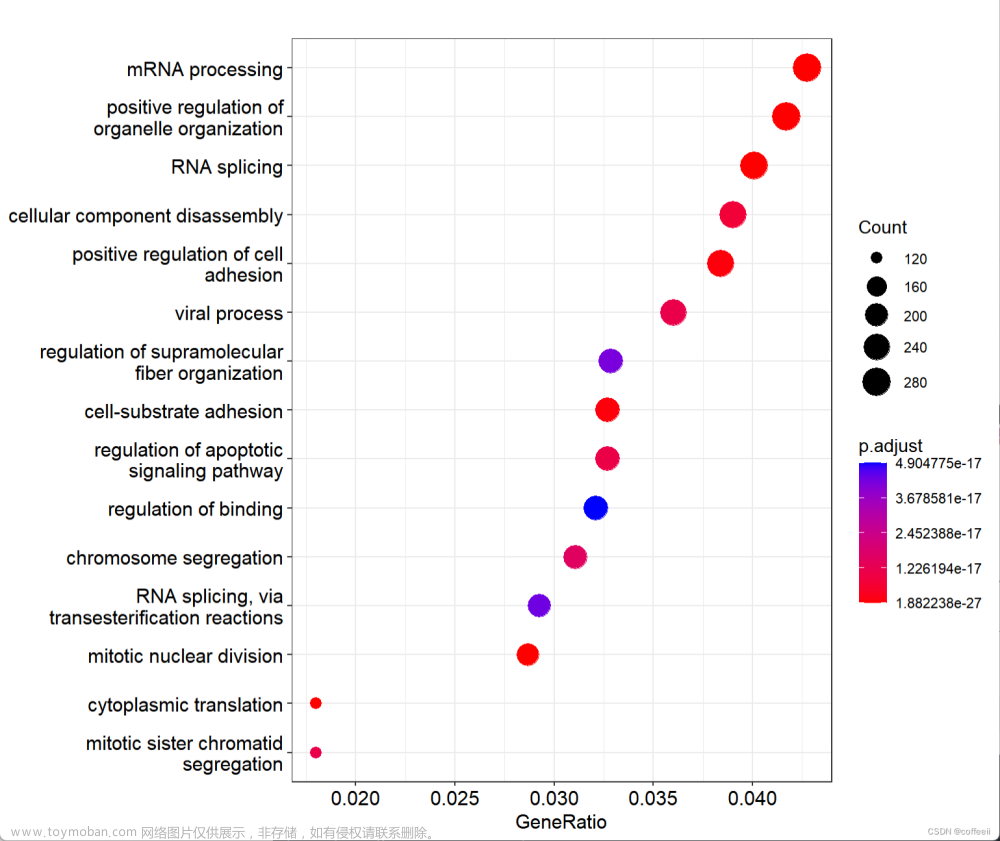
图1:横轴为富集到每个GO条目上面的基因数目

图2: 横轴为GeneRatio

图3:横轴为Fold enrichment(富集倍数)
下面我们结合富集分析的结果表,来分别解释一下这三张图中横坐标的具体含义。

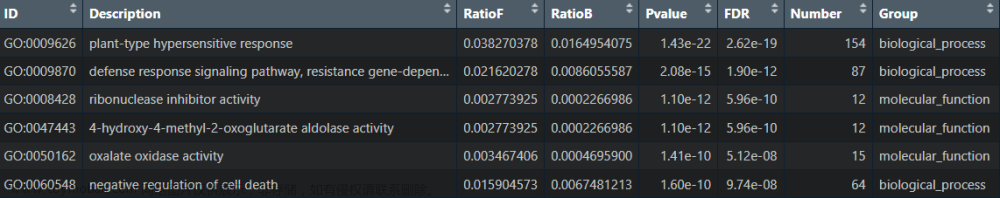
首先来看看这张表中每一列所代表的含义
ONTOLOGY:区分是BP,MF还是CC
ID:具体的GO条目的ID号
Description:GO条目的描述
GeneRatio:这里是一个分数,分子是富集到这个GO条目上的gene的数目,
分母是所有输入的做富集分析的gene的数目,可以是差异表达
分析得到的gene
BgRatio:Background Ratio. 这里也是一个分数,分母是人的所有编码蛋白的
基因中有GO注释的gene的数目,这里是19623个,分子是这19623个
gene中注释到这个GO条目上面的gene的数目
pvalue:富集的p值
p.adjust:校正之后的p值
qvalue:q值
geneID:输入的做富集分析的gene中富集到这个GO条目上面的具体的
gene名字
Count:输入的做富集分析的gene中富集到这个GO条目上面的gene的数目这张表里面没有提到富集倍数(fold enrichment)
fold enrichment = GeneRatio / BgRatio那么我们就很容易理解上面三张图的横坐标了,分别为Count,GeneRatio和Fold enrichment。
那么问题来了,既然这张表里面没有Fold enrichment,那么我们如何计算富集倍数呢?
下面小编给大家介绍三种方法来计算Fold enrichment,任君挑选
1.利用eval直接做计算
kegg=read.csv("KEGG-enrich.csv",stringsAsFactors = F)
enrichment_fold=apply(kegg,1,function(x){
GeneRatio=eval(parse(text=x["GeneRatio"]))
BgRatio=eval(parse(text=x["BgRatio"]))
enrichment_fold=round(GeneRatio/BgRatio,2)
enrichment_fold
})
2.利用strsplit按/分割成分子和分母
kegg=read.csv("KEGG-enrich.csv",stringsAsFactors = F)
fenshu2xiaoshu<-function(ratio){
sapply(ratio,function(x) as.numeric(strsplit(x,"/")[[1]][1])/as.numeric(strsplit(x,"/")[[1]][2]))
}
enrichment_fold=fenshu2xiaoshu(kegg$GeneRatio)/fenshu2xiaoshu(kegg$BgRatio)
enrichment_fold=as.numeric(enrichment_fold)
3. 利用gsub替换,得到分子和分母
kegg=read.csv("KEGG-enrich.csv",stringsAsFactors = F)
fenshu2xiaoshu2<-function(ratio){
sapply(ratio,function(x) as.numeric(gsub("/.*$","",x))/as.numeric(gsub("^.*/","",x)))
}
enrichment_fold=fenshu2xiaoshu2(kegg$GeneRatio)/fenshu2xiaoshu2(kegg$BgRatio)
enrichment_fold=as.numeric(enrichment_fold)富集分析
当分析实验过程是否显著影响某一类功能基因时,如果只是根据该类基因中差异基因的比例是不准确的。
比如说,某一类功能基因的总数是100个,其中差异基因有10个,那么只有实验只是影响了该功能基因的10%,但是如果该实验一共只鉴定到20个差异基因,那么这10个差异基因就占所有差异基因的50%,因此,需要同时考虑差异基因在所有功能分类中的分布,得到的结果才是准确的。
GO和KEGG富集分析
对差异基因进行GO功能和KEGG通路富集分析,可以识别差异基因富集的功能或代谢路径,在基因功能和代谢通路水平阐明样本间的差异。
该分析通常使用软件Goatools和KOBAS进行, 富集显著性的检验方法为Fisher精确检验。
为控制分析结果的假阳性率,还需要对检验结果的显著性进行校正,可以使用4种多重检验方法 (Bonferroni、Holm、Sidak和false discovery rate) 对p值进行了校正。
一般情况下,当经过校正的p值 (qvalue) ≤ 0.05时,认为此GO功能或KEGG通路存在显著富集情况。
Fisher精确检验
应用如下公式计算显著性p值:

其中,a为功能A中差异基因的数量,b为非差异基因的数量,c为所有功能分类中差异基因的总数量,d为非差异基因的总数量,n为识别到的所有差异基因的数目。
之后使用如下公式对p值进行校正:

其中,p为待检验GO功能或KEGG路径的p值,length(p)为所有需要检验GO功能或KEGG路径的数目,rank(p)为待检验p值在所有p值从到到底排列中的位置。
结果展示
在转录组测序结果中,可以通过散点图对差异表达基因进行富集分析。
以KEGG通路富集结果为例,此图中,KEGG富集程度通过Rich factor、qvalue和富集到此通路上的基因个数来衡量。

富集分析
横坐标是Rich factor,为该代谢路径下差异基因数目与所有注释到该路径基因数目的比值,数值越大表示富集程度越大。
前面给大家简单介绍过☞什么是Gene Ontology(GO),以及如何用一些简单易用的网页工具去做GO和KEGG富集分析
☞富集分析DAVID、Metascape、Enrichr、ClueGO
☞比DAVID强一万倍的基因注释工具
☞GO,KEGG富集分析工具——DAVID
也给大家简单的介绍过如何用R做GO富集分析,以及如何将富集结果中的gene ID转成基因名字
☞GO和KEGG富集结果如何显示基因symbol
还有计算富集倍数的三种方法
☞GO和KEGG富集倍数(Fold Enrichment)如何计算、

生信交流平台
53 次咨询
5.0
上海生命科学研究院 生物信息学博士
2623 次赞同
去咨询
今天我们来聊聊怎么来展示GO富集分析的结果,下面是一个GO富集分析的结果表。

ONTOLOGY:区分是BP,MF还是CC
ID:具体的GO条目的ID号
Description:GO条目的描述
GeneRatio:这里是一个分数,分子是富集到这个GO条目上的gene的数目,
分母是所有输入的做富集分析的gene的数目,可以是差异表达
分析得到的gene
BgRatio:Background Ratio. 这里也是一个分数,分母是人的所有编码蛋白的
基因中有GO注释的gene的数目,这里是19623个,分子是这19623个
gene中注释到这个GO条目上面的gene的数目
pvalue:富集的p值
p.adjust:校正之后的p值
qvalue:q值
geneID:输入的做富集分析的gene中富集到这个GO条目上面的具体的
gene名字
Count:输入的做富集分析的gene中富集到这个GO条目上面的gene的数目我们知道GO富集分析的结果又可以进一步划分成三个类。
BP:biological process,生物学过程。
MF:molecular function,分子功能。
CC:cellular component, 细胞成分。因此在画图的时候,我们需要将这三类给区分开来。下面我们分别用了四种不同的方式来展示GO富集分析的结果。
1.气泡图分三个框显示BP,MF和CC的富集分析结果

2. 柱形图分三个框显示BP,MF和CC的富集分析结果

3. 柱形图用三种不同颜色显示BP,MF和CC的富集分析结果

4. 气泡图+标签,显示BP,MF和CC的富集分析结果

这张图的横轴是富集倍数(fold enrichment),计算方法如下
fold enrichment = GeneRatio / BgRatio上面四张图的横坐标分别为GeneRatio,Count,−log10(p.adjust)和Fold enrichment。文章来源:https://www.toymoban.com/news/detail-738364.html
☞完整的GO/KEGG富集分析及绘图代码的视频讲解文章来源地址https://www.toymoban.com/news/detail-738364.html
到了这里,关于☞GO和KEGG富集倍数(Fold Enrichment)如何计算 enrich factor qvalue的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!