上期“干货预警——原来基因功能富集分析这么简单!”和“【R语言】——基因GO/KEGG功能富集结果可视化(保姆级教程)”介绍如何使用DAVID在线分析工具对基因进行GO/KEGG功能富集分析和使用R ggplot包对获得的基因GO/KEGG功能富集结果进行可视化。本期介绍使用R clusterProfiler包和R AnnotationHub包对基因进行GO/KEGG功能富集分析、OrgDb包制作以及结果可视化。
GO/KEGG功能富集分析中重要的是背景基因的选择,使用R clusterProfiler包对基因进行富集,需要导入目的基因(前景基因)相对应物种的参考基因组(背景基因),现阶段“bioconductor”已有十几种常见动物,如人类、小鼠等物种的OrgDb。但仍然有许多物种不在Bioconductor的OrgDb列表里,但存在参考基因组,如山羊,绵羊等,这种情况则需要用到R AnnotationHub包进行索引其对应物种的参考基因组,并制作OrgDb包使用。
1 数据准备
数据输入格式(xlsx格式):

2 R包加载、数据导入及处理
#下载包#
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
BiocManager::install("topGO")
BiocManager::install("Rgraphviz")
BiocManager::install("pathview")
install.packages("ggplot2")
BiocManager::install('stringr')
install.packages("openxlsx")
#加载包#
library(clusterProfiler)
library(topGO)
library(Rgraphviz)
library(pathview)
library(ggplot2)
library(stringr)
library(openxlsx)
#导入数据#
remove(list = ls()) #清除 Global Environment
getwd() #查看当前工作路径
setwd("C:/Rdata/jc") #设置需要的工作路径
list.files() #查看当前工作目录下的文件
data = read.xlsx("enrich-gene.xlsx",sheet= "enrich_genes",sep=',') #导入数据
head(data)
#数据处理-差异基因筛选#
vector = abs(data$log2FC) > 1 & data$PValue < 0.05 & data$gene_name !="" ##abs绝对值;通常logFC> 1和PValue< 0.05条件进行筛选;data$gene_name != ""表示gene_name不为空白
#data$gene_name<-str_to_title(data$gene_name)#用stringr将基因名称的第一个字母大写(小鼠首字母为大写)
data_sgni= data[vector,]#筛选差异基因
head(data_sgni)
#All_gene <- rownames(data) # 提取所有基因基因名
3 背景基因选择及GO/KEGG富集分析
3.1 在“bioconductor”中已有OrgDb的物种的富集分析
在http://bioconductor.org/packages/release/BiocViews.html#___OrgDb 中寻找需要物种的OrgDb(目前仅有下图所示物种),以人类“org.Hs.eg.db”为例:

图1 “bioconductor”中已有OrgDb的物种
#已有OrgDb的常见物种#
BiocManager::install("org.Hs.eg.db")
library(org.Hs.eg.db)
#基因ID转换#
keytypes(org.Hs.eg.db) #查看所有可转化类型
entrezid_all = mapIds(x = org.Hs.eg.db, #id转换的比对基因组(背景基因)的物种,以人为例
keys = data_sgni$gene_name, #将输入的gene_name列进行数据转换
keytype = "SYMBOL", #输入数据的类型
column = "ENTREZID")#输出数据的类型
entrezid_all = na.omit(entrezid_all) #na省略entrezid_all中不是一一对应的数据情况
entrezid_all = data.frame(entrezid_all) #将entrezid_all变成数据框格式
head(entrezid_all)
###GO富集分析###
GO_enrich = enrichGO(gene = entrezid_all[,1], #表示前景基因,即待富集的基因列表;[,1]表示对entrezid_all数据集的第1列进行处理
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID", #输入数据的类型
ont = "ALL", #可以输入CC/MF/BP/ALL
#universe = 背景数据集 # 表示背景基因,无参的物种选择组装出来的全部unigenes作为背景基因;有参背景基因则不需要。
pvalueCutoff = 1,qvalueCutoff = 1, #表示筛选的阈值,阈值设置太严格可导致筛选不到基因。可指定 1 以输出全部
readable = T) #是否将基因ID映射到基因名称。
GO_enrich = data.frame(GO_enrich) #将GO_enrich导成数据框格式
#数据导出#
write.csv(GO_enrich,'C:/Rdata/保存文件/GO_enrich.csv') ###KEGG富集分析###
KEGG_enrich = enrichKEGG(gene = entrezid_all[,1], #即待富集的基因列表
keyType = "kegg",
pAdjustMethod = 'fdr', #指定p值校正方法
organism= "human", #hsa,可根据你自己要研究的物种更改,可在https://www.kegg.jp/brite/br08611中寻找
qvalueCutoff = 1, #指定 p 值阈值(可指定 1 以输出全部)
pvalueCutoff=1) #指定 q 值阈值(可指定 1 以输出全部)
KEGG_enrich = data.frame(KEGG_enrich)
write.csv(KEGG_enrich,'C:/Rdata/保存文件/KEGG_enrich.csv') #数据导出3.2 使用“AnnotationHub”获取在线注释并创建OrgDb对象
如果你所研究的物种不在Bioconductor的OrgDb列表里,但存在参考基因组,如山羊(Capra hircus/goat/chx),绵羊(sheet/Ovis aries)等,这种情况则需要用到AnnotationHub函数进行索引其对应物种的参考基因组(背景基因),并制作OrgDb包使用。
注意:AnnotationHub包连接的Bioconductor数据库是实时更新的,所以需要用到的时候再在线查询和使用。
###制作可索引到物种的OrgDb包###
#下载和加载包#
BiocManager::install("AnnotationHub")
BiocManager::install("AnnotationDbi")
BiocManager::install("rtracklayer")
library(AnnotationHub)
library(AnnotationDbi)
library(rtracklayer)
#索引与制作OrgDb#
hub <- AnnotationHub() #建立AnnotationHub对象保存到hub
query(hub, 'Capra hircus') #查询包含山羊(Capra hircus)的物种信息;结果有物种的各类信息需要进一步筛选
query(hub[hub$rdataclass == "OrgDb"] , "Capra hircus") #筛选我们需要OrgDb类型;也可将上一步与这一步合并成query(hub,'org.Capra hircus')进行搜索
goat <- hub[['AH101444']] #制作Capra hircus的OrgDb库;AH101444是Capra hircus对应的编号。
goat #查看goat
#help('select') 
#保存、载入与查看-AnnotationDbi#
saveDb(goat,file="goat.OrgDb") #把goat对象保存成goat.OrgDb文件
goat = loadDb(file="goat.OrgDb") #载入goat.OrgDb文件,保存到goat
length(keys(goat)) #查看包含的基因数量
columns(goat) #查看goat的数据类型
keys(goat, keytype = "SYMBOL") #查看SYMBOL数据集下的ID
# 查看AnnotationHub内容——根据自己兴趣了解#
#display(hub) #调出网页
#unique(hub$species) #查看hub里包含的所有物种
#unique(hub$rdataclass) #查看hub里的数据类型
#hub[hub$rdataclass == "OrgDb"] #查看hub里OrgDb类型的数据#基因ID转换#
keytypes(goat) #查看所有可转化类型
entrezid_all = mapIds(x = goat, #id转换的比对基因组(背景基因)所属物种,这边为山羊
keys = data_sgni$gene_name, #将输入的gene_name列进行数据转换
keytype = "SYMBOL", #输入数据的类型
column = "ENTREZID")#输出数据的类型
entrezid_all = na.omit(entrezid_all) #na省略entrezid_all中不是一一对应的数据情况
entrezid_all = data.frame(entrezid_all) #将entrezid_all变成数据框格式
head(entrezid_all)
#GO富集分析#
GO_enrich = enrichGO(gene = entrezid_all[,1], #待富集的基因列表
OrgDb = goat, #指定物种的基因数据库,goat直接赋值给OrgDb参数即可
keyType = 'ENTREZID', #输入数据的类型
ont = 'ALL', #可指定 BP\MF\CC\ALL
pAdjustMethod = 'fdr', #指定 p 值校正方法
pvalueCutoff = 1, #指定 p 值阈值(指定 1 以输出全部)
qvalueCutoff = 1, #指定 q 值阈值(指定 1 以输出全部)
readable = FALSE)
GO_enrich = data.frame(GO_enrich)
write.csv(GO_enrich,'C:/Rdata/保存文件/GO_enrich.csv') #数据导出#
###KEGG富集分析###
KEGG_enrich = enrichKEGG(gene = entrezid_all[,1], #即待富集的基因列表
keyType = "kegg",
pAdjustMethod = 'fdr', #指定p值校正方法
organism= "chx", #山羊,可根据你自己要研究的物种更改,可在https://www.kegg.jp/brite/br08611中寻找
qvalueCutoff = 1, #指定 p 值阈值(可指定 1 以输出全部)
pvalueCutoff=1) #指定 q 值阈值(可指定 1 以输出全部)
KEGG_enrich = data.frame(KEGG_enrich)
write.csv(KEGG_enrich,'C:/Rdata/保存文件/KEGG_enrich.csv') #数据导出

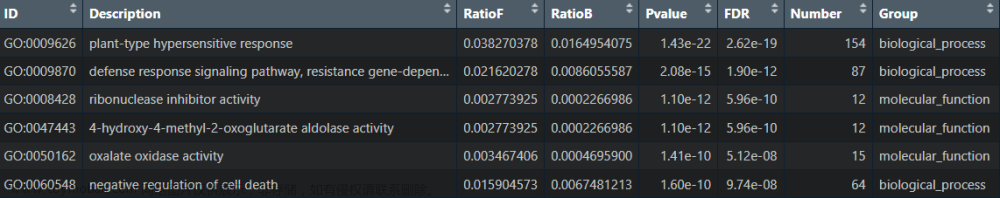
输出的GO/KEGG富集结果各列内容:
ONTOLOGY:GO的BP(生物学过程)、CC(细胞组分)或MF(分子功能)三个方面内容;
ID:富集到的GO term/KEGG term;
Description:对GO term/KEGG term的生物学功能和意义进行描述;
GeneRatio:富集到该GO term/KEGG term中的基因数目/给定基因的总数目;
BgRatio:该GO term/KEGG term中背景基因总数目/该物种所有已知GO功能基因的数目;
pvalue、p.adjust和qvalue:p值、校正后p值和q值;
geneID和Count:富集到该GO term/KEGG term中的基因名称和数目。
4 GO/KEGG富集结果可视化
###GO/KEGG富集结果可视化###
#数据载入与处理#
install.packages("ggplot2")
library(ggplot2)
go_enrich = read.xlsx("enrich-gene.xlsx",sheet= "ONTOLOGY",sep=',')
go_enrich$term <- paste(go_enrich$ID, go_enrich$Description, sep = ': ') #将ID与Description合并成新的一列
go_enrich$term <- factor(go_enrich$term, levels = go_enrich$term,ordered = T)
#纵向柱状图#
ggplot(go_enrich,
aes(x=term,y=Count, fill=ONTOLOGY)) + #x、y轴定义;根据ONTOLOGY填充颜色
geom_bar(stat="identity", width=0.8) + #柱状图宽度
scale_fill_manual(values = c("#6666FF", "#33CC33", "#FF6666") ) + #柱状图填充颜色
facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')+
coord_flip() + #让柱状图变为纵向
xlab("GO term") + #x轴标签
ylab("Gene_Number") + #y轴标签
labs(title = "GO Terms Enrich")+ #设置标题
theme_bw()
#help(theme) #查阅这个函数其他具体格式#横向柱状图#
ggplot(go_enrich,
aes(x=term,y=Count, fill=ONTOLOGY)) + #x、y轴定义;根据ONTOLOGY填充颜色
geom_bar(stat="identity", width=0.8) + #柱状图宽度
scale_fill_manual(values = c("#6666FF", "#33CC33", "#FF6666") ) + #柱状图填充颜色
facet_grid(.~ONTOLOGY, scale = 'free_x', space = 'free_x')+
xlab("GO term") + #x轴标签
ylab("Gene_Number") + #y轴标签
labs(title = "GO Terms Enrich")+ #设置标题
theme_bw() +
theme(axis.text.x=element_text(family="sans",face = "bold", color="gray50",angle = 70,vjust = 1, hjust = 1 )) #对字体样式、颜色、还有横坐标角度()#气泡图#
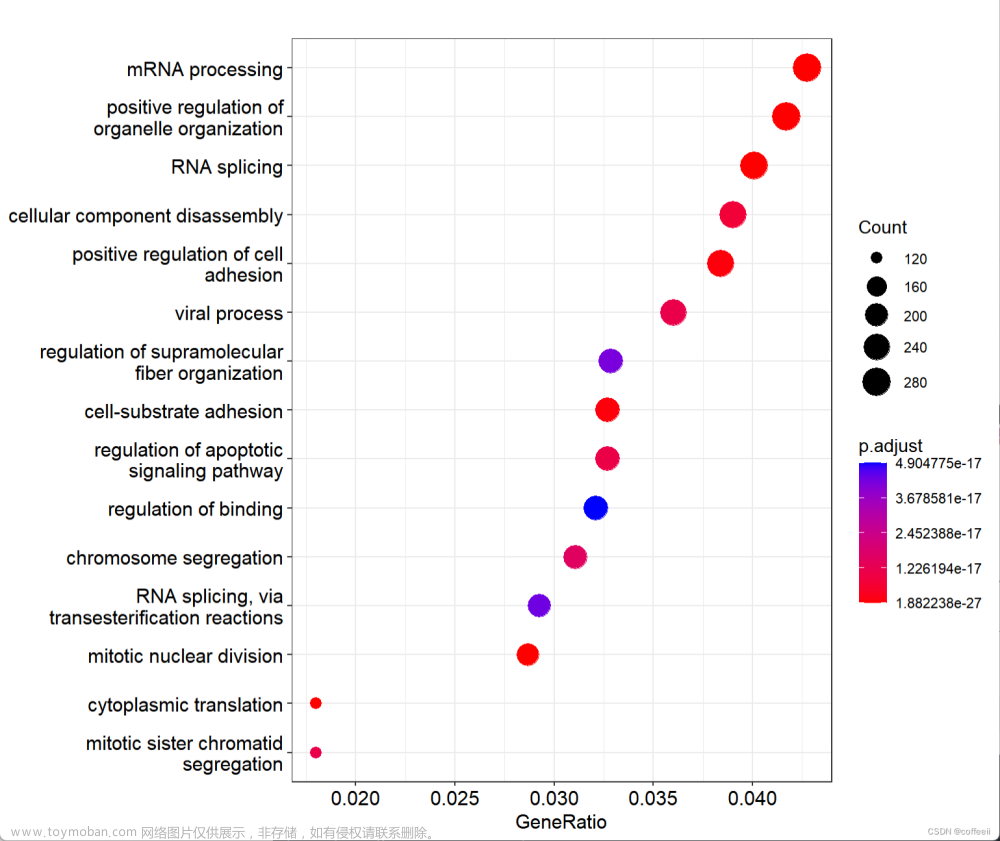
ggplot(go_enrich,
aes(y=term,x=Count))+
geom_point(aes(size=Count,color=p.adjust))+
facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')+
scale_color_gradient(low = "red",high ="blue")+
labs(color=expression(PValue,size="Count"),
x="Gene Ratio",y="GO term",title="GO Enrichment")+
theme_bw()


图1 为GO富集结果图
KEGG富集结果与GO富集结果可视化类似可参考上一期“【R语言】——基因GO/KEGG功能富集结果可视化(保姆级教程)”内容。
好了本次分享就到这里,下期有更精彩内容,敬请期待。文章来源:https://www.toymoban.com/news/detail-786452.html
关注“在打豆豆的小潘学长”公众号,发送“富集分析2”获得完整代码包和演示数据。文章来源地址https://www.toymoban.com/news/detail-786452.html
到了这里,关于【R语言】——基因GO/KEGG富集分析!超级简单的保姆级教程!的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!