生信数据分析——GO+KEGG富集分析
1. 富集分析基础知识
1.1 为什么要做功能富集分析?
转录组学数据得到的基因非常多,面对大量的基因无法做到挨个研究其功能,因此为了研究基因所具有的功能,将部分功能相似的基因进行归类,这样具有相似功能的基因就被放在一起,构成了一个通路,从而减少工作量,并可以实现功能和表型相关联。
1.2 什么是富集分析?
富集分析是一种数据分析方法,主要用于理解基因集合或其他生物学实体在特定实验条件或生物学背景下的功能、通路或特定生物学过程的富集程度。其基本原理是,如果某个基因集合在特定条件下显著富集于某个功能类别或通路中,那么这些基因可能共同参与了某种特定的生物学过程或具有某种共同的功能特性
看上方的描述是不是感觉晦涩难懂,简单地说:所谓富集分析,本质上就是对分布的检验,如果基因分布集中在某一个区域(通路),则认为富集。
举个栗子: 做完差异后,得到了一堆差异基因,现在对这部分差异基因归归类,部分功能相似的基因可能被划分到了炎症通路上,有的基因被划分到了代谢通路上,这样就能大致知道筛选出来的差异基因与哪些功能相关。
1.3 富集分析有几种类型?
(1)GO富集分析
GO富集分析会从三个方面描述基因潜在的功能,分别是:
- 分子功能(Molecular Function,MF)——即基因是否富集到分子相关的通路上
- 细胞组分(Cellular Component,CC)——即基因定位在细胞的哪个位置上
- 参与的生物过程(Biological Process,BP)——即基因参与哪些生物学过程
举个栗子:离子通道活性的GO term是GO:0005216,如果差异基因富集到该term上,那么所研究的基因可能与离子通道的激活与抑制有关联。
(2)KEGG富集分析
京都基因与基因组百科全书(KEGG)是了解高级功能和生物系统(如细胞、生物和生态系统)、用于研究通路的数据库之一。KEGG 通路分析是借助 KEGG 数据库(Kyoto Encyclopedia of Genes and Genomes),对所有鉴定到的基因进行通路注释,并分析这些基因参与的主要代谢和信号转导途径。
简单说: 使用KEGG数据库中通路的注释信息,将基因与已知的代谢通路和功能进行关联
(3)GSEA富集分析
(4)GSVA富集分析
在这个分析点中重点关注GO富集分析和KEGG富集分析,GSEA和GSVA会在后面分析点中介绍。
2. GO富集分析(Rstudio)
本项目以 ADAMTS2, ADAMTS4, AGRN, COL5A1, CTSB, FMOD, LAMB3, LAMB4, LOXL2, MATN1, MEP1A, MMP1, MMP2, NTN1, PTN, SPARCL1, SPON1, TGFBI, THBS4, TNC, VTN, ITGB6, PTPRF, UNC5A 为例展示GO富集分析过程
物种:人类(Homo sapiens)
R版本:4.2.2
R包:tidyverse,clusterProfiler,org.Hs.eg.db
废话不多说,代码如下:
设置工作空间:
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./02_GO+KEGG_enrichment')){
dir.create('./02_GO+KEGG_enrichment')
} # 判断该工作路径下是否存在名为02_GO+KEGG_enrichment的文件夹,如果不存在则创建,如果存在则pass
setwd('./02_GO+KEGG_enrichment/') # 设置路径到刚才新建的02_GO+KEGG_enrichment下
加载包:
library(clusterProfiler)
library(org.Hs.eg.db)
library(tidyverse)
导入要富集分析的基因
gene <- c('ADAMTS2', 'ADAMTS4', 'AGRN', 'COL5A1', 'CTSB', 'FMOD', 'LAMB3', 'LAMB4', 'LOXL2',
'MATN1', 'MEP1A', 'MMP1', 'MMP2', 'NTN1', 'PTN', 'SPARCL1', 'SPON1', 'TGFBI', 'THBS4', 'TNC',
'VTN', 'ITGB6', 'PTPRF', 'UNC5A')
设置数据库(注意:由于本项目分析的是人类基因,因此选用的是org.Hs.eg.db,如果是其他物种,需要用其他数据库)
GO_database <- 'org.Hs.eg.db' # GO是org.Hs.eg.db数据库
gene ID转换(因为导入的是基因名(symbol),但是用官方的编号,也就是ENTREZID会比较专业一些,因此首先要将基因名转换成官方ENTREZID)
gene <- bitr(gene, fromType = 'SYMBOL', toType = 'ENTREZID', OrgDb = GO_database)
知识拓展: bitr函数不仅能将symbol转成ENTREZID,还能将ENTREZID转回symbol,甚至还能转换成其他形式,具体可以自行查看官方说明!
gene 如下图所示,第一列就是基因名(symbol),而第二列就是官方的ENTREZID编号
(注意:用bitr做转换的时候,很有可能会出现基因没有对应的ENTREZID编号,这是一个正常现象,不用过多焦虑,合理解释就行!)
GO富集分析并将富集分析结果转成数据框,enrichGO函数常用参数介绍如下:
- gene参数——是要输入的基因(一般用基因的ENTREZID编号)
- OrgDb 参数——指定要用到的数据库,人类是:org.Hs.eg.db(当然还有别的物种,可自行查询)
- keyType参数——设定读取的gene ID类型,本教程用的是ENTREZID编号所以用“ENTREZID”
- ont参数——指定输出的通路类型,前面也说了GO富集分析会从bp,cc,mf三个层次描述基因的功能,这里用ALL就会直接包括这三个部分,当然也可以只指定一种类型。
- pvalueCutoff 参数——设定p值阈值
- qvalueCutoff 参数——设定q值阈值(这个q值就是矫正后的p值)
- readable参数——当readable设置为TRUE时,函数的输出会以一种更易于阅读和理解的方式呈现
(enrichGO函数中比较关注的参数就是上述的这些,当然还有其他参数,如果想深入了解可自行查看官方说明文档)
GO <- enrichGO(gene = gene$ENTREZID, # 导入基因的ENTREZID编号
OrgDb = GO_database, # 用到的数据库(人类是:org.Hs.eg.db)
keyType = "ENTREZID", # 设定读取的gene ID类型
ont = "ALL", # (ont为ALL因此包括 Biological Process,Cellular Component,Mollecular Function三部分)
pvalueCutoff = 0.5, # 设定p值阈值
qvalueCutoff = 0.5, # 设定q值阈值
readable = T)
go_res <- data.frame(GO) # 将GO结果转为数据框
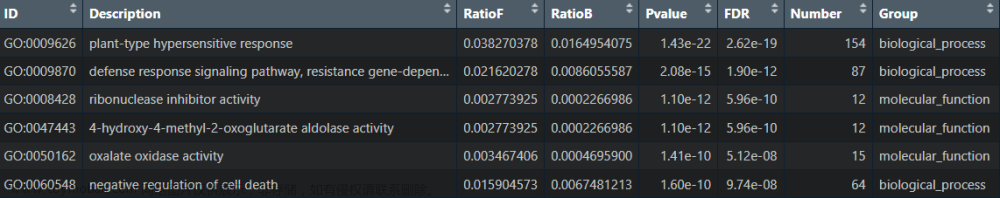
go_res 如下图所示:
- ONTOLOGY——指示该通路属于哪个类别,即生物过程(Biological Process, BP)、分子功能(Molecular Function, MF)还是细胞组分(Cellular Component, CC)
- ID——这是GO通路的唯一标识符,用于在GO数据库中唯一地标识一个通路(可以理解成身份证)
- Description——对通路的简单描述,通常通过这一列就得知该通路具有哪些功能
- GeneRatio——是富集到该通路上的基因数量与所有输入到富集分析中的基因数量的比值。它反映了在特定基因集合中,与该通路相关的基因所占的比例。
- BgRatio——是在整个背景数据集(通常是整个基因组或某个参考数据集)中,与该通路相关的基因数量与背景数据集中所有基因数量的比值。它反映了在整个基因组中,与该通路相关的基因所占的比例。
- pvalue,p.adjust,qvalue——都是GO富集结果的显著性pvalue是常规p值,另外两个是调整后的p值,通常只需要pvalue < 0.05即可。
- geneID——是富集到该通路上的基因名
- Count——是富集到该通路上的基因数目

给go_res 添加新的一列——richFactor
- RichFactor——是一个重要的指标,用于衡量差异表达的转录本中位于特定通路的转录本数目与所有有注释转录本中位于该通路的转录本总数的比值。
简单说:RichFactor越大,表示富集的程度越大,其评价富集的效果要比单纯的GeneRatio或Count要好
go_res <- mutate(go_res, richFactor = Count / as.numeric(sub("/\\d+", "", BgRatio)))
最后筛选p值显著的通路,并保存结果
go_res <- go_res[go_res$pvalue<0.05, ]
write.csv(go_res, file = "./GO_res.csv")
3. KEGG富集分析(Rstudio)
分析与GO类似,这里同样是从头开始展示
本项目以 ADAMTS2, ADAMTS4, AGRN, COL5A1, CTSB, FMOD, LAMB3, LAMB4, LOXL2, MATN1, MEP1A, MMP1, MMP2, NTN1, PTN, SPARCL1, SPON1, TGFBI, THBS4, TNC, VTN, ITGB6, PTPRF, UNC5A 为例展示GO富集分析过程
物种:人类(Homo sapiens)
R版本:4.2.2
R包:tidyverse,clusterProfiler,org.Hs.eg.db
设置工作空间:
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./02_GO+KEGG_enrichment')){
dir.create('./02_GO+KEGG_enrichment')
} # 判断该工作路径下是否存在名为02_GO+KEGG_enrichment的文件夹,如果不存在则创建,如果存在则pass
setwd('./02_GO+KEGG_enrichment/') # 设置路径到刚才新建的02_GO+KEGG_enrichment下
加载包:
library(clusterProfiler)
library(org.Hs.eg.db)
library(tidyverse)
导入要富集分析的基因
gene <- c('ADAMTS2', 'ADAMTS4', 'AGRN', 'COL5A1', 'CTSB', 'FMOD', 'LAMB3', 'LAMB4', 'LOXL2',
'MATN1', 'MEP1A', 'MMP1', 'MMP2', 'NTN1', 'PTN', 'SPARCL1', 'SPON1', 'TGFBI', 'THBS4', 'TNC',
'VTN', 'ITGB6', 'PTPRF', 'UNC5A')
设置数据库(注意:这里和前面区别就在于要指定KEGG数据库,即hsa(人种))
GO_database <- 'org.Hs.eg.db' # GO是org.Hs.eg.db数据库
KEGG_database <- 'hsa' # KEGG是hsa数据库
同样是gene ID转换
gene <- bitr(gene, fromType = 'SYMBOL', toType = 'ENTREZID', OrgDb = GO_database)
gene 如下图所示,第一列就是基因名(symbol),而第二列就是官方的ENTREZID编号
接下来就是KEGG富集分析,enrichGO函数常用参数介绍如下
- gene参数——是要输入的基因(一般用基因的ENTREZID编号)
- keyType参数——指定了基因ID的类型,用于匹配KEGG数据库中的条目
- organism参数——指定了进行富集分析的目标物种的KEGG数据库,由于基因用的是人类的,所以前面设置的“hsa”。
- pAdjustMethod参数——指定了用于调整p值的统计方法,以控制假阳性率
- pvalueCutoff 参数——设定p值阈值
- qvalueCutoff 参数——设定q值阈值(这个q值就是矫正后的p值)
KEGG <- enrichKEGG(gene = gene$ENTREZID,
keyType = "kegg",
organism = KEGG_database,
pAdjustMethod = "BH",
pvalueCutoff = 0.5,
qvalueCutoff = 0.5)
KEGG 如下图所示,是一个列表,里面在这里比较重要的是gene那里,可以看到那里不是常规的基因名,因此不能直接将KEGG的结果转换成数据框,多了一个基因ID转换的过程。
将KEGG结果中基因ID转成基因名,之后将KEGG结果转成数据框
kegg_res <- setReadable(KEGG, OrgDb = org.Hs.eg.db, keyType="ENTREZID")
kegg_res <- data.frame(kegg_res)
kegg_res 结果如下图所示:
- ID——这是KEGG通路的唯一标识符,用于在KEGG数据库中唯一地标识一个通路(可以理解成身份证)
- Description——对通路的简单描述,通常通过这一列就得知该通路具有哪些功能
- GeneRatio——是富集到该通路上的基因数量与所有输入到富集分析中的基因数量的比值。它反映了在特定基因集合中,与该通路相关的基因所占的比例。
- BgRatio——是在整个背景数据集(通常是整个基因组或某个参考数据集)中,与该通路相关的基因数量与背景数据集中所有基因数量的比值。它反映了在整个基因组中,与该通路相关的基因所占的比例。
- pvalue,p.adjust,qvalue——都是GO富集结果的显著性pvalue是常规p值,另外两个是调整后的p值,通常只需要pvalue < 0.05即可。
- geneID——是富集到该通路上的基因名
- Count——是富集到该通路上的基因数目

同样给kegg_res 添加新的一列——richFactor
kegg_res <- mutate(kegg_res , richFactor = Count / as.numeric(sub("/\\d+", "", BgRatio)))
最后筛选p值显著的通路,并保存结果
kegg_res <- kegg_res [kegg_res $pvalue<0.05, ]
write.csv(kegg_res , file = "./KEGG_res.csv")
结语:
以上就是GO+KEGG富集分析的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。文章来源:https://www.toymoban.com/news/detail-848510.html
如果觉得本教程对你有所帮助,点赞关注不迷路!!!文章来源地址https://www.toymoban.com/news/detail-848510.html
- 目录部分跳转链接:零基础入门生信数据分析——导读
到了这里,关于生信数据分析——GO+KEGG富集分析的文章就介绍完了。如果您还想了解更多内容,请在右上角搜索TOY模板网以前的文章或继续浏览下面的相关文章,希望大家以后多多支持TOY模板网!







![[数据挖掘、数据分析] clickhouse在go语言里的实践](https://imgs.yssmx.com/Uploads/2024/02/714916-1.png)